












Μεγάλη θλίψη στον καλλιτεχνικό – και όχι μόνο – κόσμο σκόρπισε ο θάνατος του αγαπημένου ηθοποιού Γεράσιμου Μιχελή που έφυγε χθες από τη ζωή σε ηλικία 60 ετών.
Ο ηθοποιός όπως έγινε γνωστό έπασχε από τη λεγόμενη «νόσο του κινητικού νευρώνα» – γνωστή και ως αμυοτροφική πλάγια σκλήρυνση.
Τι ακριβώς είναι η νόσος του κινητικού νευρώνα
Η ALS (πλαγία αμυοτροφική σκλήρυνση ή νόσος του κινητικού νευρώνα) είναι μια νευροεκφυλιστική πάθηση που επηρεάζει τους κινητικούς νευρώνες του Κεντρικού και Περιφερικού Νευρικού Συστήματος, με συνέπεια την προϊούσα μυϊκή ατροφία. Ξεκινάει με ένα μούδιασμα και αδυναμία σε κάποιο άκρο και καταλήγει σε κινητική παράλυση χεριών ή και ποδιών, αδυναμία κατάποσης και αναπνευστική ανεπάρκεια.
Η διάγνωση της νόσου γίνεται από το ιστορικό και την κλινική εξέταση και επιβεβαιώνεται από το ηλεκτρομυογράφημα. Δεν υπάρχει μέχρι τώρα οριστική θεραπεία της νόσου. Για να επιβραδυνθεί η πορεία της χορηγούνται ειδικά φάρμακα και γίνεται εντατική φυσικοθεραπεία.
Η πάθηση αυτή είναι γνωστή και ως αμυοτροφική πλάγια σκλήρυνση.
Η παλαιότερη γνωστή περιγραφή της νόσου έγινε το 1824 από τον Τσαρλς Μπελ. Το 1869 ο Ζαν-Μαρτέν Σαρκό (Jean-Martin Charcot) συσχέτισε πρώτος τα συμπτώματα με τα νευρολογικά προβλήματα και το 1874 χρησιμοποιήθηκε για πρώτη φορά ο όρος πλαγία μυατροφική σκλήρυνση, ενώ ενίοτε αναφέρεται και ως Νόσος Σαρκό.
Η νόσος απέκτησε ευρύτερη δημοσιότητα στις ΗΠΑ το 1939, όταν διαγνώστηκε με αυτήν ο αθλητής του μπέιζμπολ Λου Γκέρινγκ (έτσι η νόσος μνημονεύεται και ως Νόσος Λου Γκέρινγκ), ενώ προσέλκυσε αργότερα διεθνές ενδιαφέρον, όταν έγινε γνωστό ότι έπασχε από την νόσο ο διάσημος επιστήμονας Στήβεν Χόκινγκ.
Τα αίτια που προκαλούν την ALS είναι άγνωστα. Ένα σημαντικό βήμα προς την απάντηση σε αυτή την ερώτηση έγινε το 1993 όταν οι επιστήμονες ανακάλυψαν ότι μεταλλάξεις στο γονίδιο που παράγει την υπεροξειδική δυσμουτάση (Cu/Zn superoxide dismutase (SOD1) σχετίζεται άμεσα με την κληρονομική μορφή της ALS. Αυτό το ένζυμο είναι ένα δυνατό αντι-οξειδωτικό που προστατεύει το σώμα από την ζημιά που προκαλεί το υπεροξείδιο, μια τοξική ελευθέρων ρίζα οξυγόνου (free radical).
Πόσο συχνή είναι η ALS ;
Η ALS είναι η 3η σε συχνότητα νευροεκφυλιστική διαταραχή, μετά το Alzheimer και το Parkinson. Εμφανίζεται σε συχνότητα 1 στα 100.000 άτομα κάθε χρόνο. Ο κίνδυνος είναι μεγαλύτερος στους άνδρες και στις λευκές φυλές. Η κύρια ηλικία εμφάνισης είναι τα 50 με 75 έτη, ενώ μετά τα 80 έτη ο κίνδυνος μειώνεται δραματικά.
Ποιες μορφές έχει η νόσος του κινητικού νευρώνα ;
Υπάρχει η σποραδική μορφή που περιλαμβάνει το 95% των περιπτώσεων και η κληρονομική μορφή (5%), όταν ανευρίσκονται πάνω από 1 περιστατικά ALS μέσα σε μια οικογένεια. Μια άλλη ταξινόμηση της ALS είναι:
– limb-onset ALS: ξεκινάει με αδυναμία στα χέρια και στα πόδια
– bulbar-onset ALS: ξεκινάει με διαταραχές του λόγου, της μάσησης και της κατάποσης.
– respiratory-onset ALS: ξεκινάει με δύσπνοια και ορθόπνοια. Έχει την χειρότερη πρόγνωση.
Επιπλέον, υπάρχουν παραλλαγές της ALS, στις οποίες πλήττεται μόνο μια συγκεκριμένη ανατομική περιοχή. Αυτές είναι:
– Αμυοτροφική διπληγία άνω άκρων: Παρουσιάζεται στους νευρώνες που ελέγχουν την κίνηση του χεριού, με αποτέλεσμα αδυναμία και παράλυση των άνω άκρων
– Αμυοτροφική διπληγία κάτω άκρων: Παρουσιάζεται στους νευρώνες που ελέγχουν την κίνηση των κάτω άκρων, με αποτέλεσμα αδυναμία και παράλυση των κάτω άκρων
– προμηκική ALS: Τα συμπτώματα είναι δυσαρθρία, δυσκαταποσία και δύσπνοια.
Τέλος, η ALS ταξινομείται ανάλογα με την ηλικία εμφάνισης :
– Young-onset ALS: Εμφανίζεται πριν τα 42 έτη, συχνότερα σε άντρες, τα κύρια συμπτώματα είναι δυσαρθρία και δυσκαταποσία και έχει πιο αργή εξέλιξη.
– Juvenile-onset ALS: Εμφανίζεται πριν τα 25 και είναι συνήθως κληρονομικού τύπου.
– Late-onset ALS: Εμφανίζεται μετά τα 65 έτη και έχει γρήγορη εξέλιξη και κακή πρόγνωση.
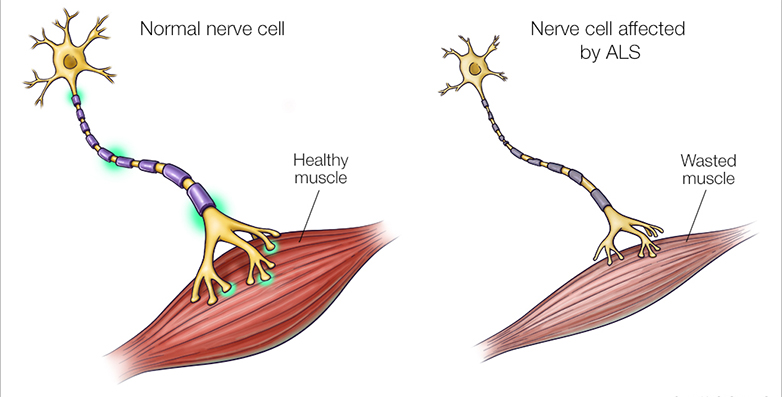
Aριστερά μια φυσιολογική νευρομυϊκή σύναψη και δεξιά μια νευρομυϊκή σύναψη σε νόσο του κινητικού νευρώνα
Ποια είναι τα συμπτώματα της ALS ;
Τα κύρια συμπτώματα είναι:
– Μυϊκή αδυναμία και παράλυση: Αδυναμία στα χέρια και στα πόδια, ατροφία μυών μάσησης και κατάποσης, αδυναμία στήριξης του κορμού και του κεφαλιού και τέλος, ατροφία των αναπνευστικών μυών
– Μυϊκές κράμπες
– Νοητικές και συμπεριφορικές διαταραχές (30-50% των ασθενών): απώλεια μνήμης, αφασία και αντικοινωνική συμπεριφορά
– Πόνος: με μορφή νευροπαθητικού πόνου, σπαστικότητας, μυϊκών κραμπών, αλλά και λόγω κατάκλισης.
Να τονιστεί πως στην πλειοψηφία των ατόμων με ALS η όραση, η ακοή, η όσφρηση, η αφή και η γεύση διατηρούνται.
Αν και ακόμα δεν είναι καθαρό το πώς η μετάλλαξη στο γονίδιο SOD1 οδηγεί στην καταστροφή των κινητικών νευρώνων, οι ερευνητές θεωρούν ότι μια συσσώρευση της ελευθέρης ρίζας οξυγόνου μπορεί να προκληθεί από την λανθασμένη λειτουργία αυτού του γονιδίου.
Επίσης, μελέτες έχουν επικεντρωθεί στον ρόλο του γλουταμικού οξέως (glutamate) στην καταστροφή των κινητικών νευρώνων. Το γλουταμικό οξύ είναι ένας από τους χημικούς μεταφορείς σημάτων ή νευροδιαβιβαστές στον εγκέφαλο. Οι επιστήμονες έχουν ανακαλύψει ότι, σε σχέση με τους υγιείς ανθρώπους, οι ασθενείς με ALS έχουν αυξημένα επίπεδα γλουταμικού οξέως στον ορό του αίματος και στο εγκεφαλονωτιαίο υγρό.
Τα γονίδια που ευθύνονται για την οικογενή μορφή της ALS έχουν ταυτοποιηθεί σε ποσοστό >50% των οικογενών περιπτώσεων ALS.
Μεταξύ αυτών, μεταλλαγή στο γονίδιο SOD1 ευθύνεται για το 20% των οικογενών περιπτώσεων ALS, μεταλλαγές στα γονίδια TARDBP και FUS ευθύνονται για το 4–5% των οικογενών περιπτώσεων ALS, >30% των οικογενών περιπτώσεων σχετίζονται με μεταλλαγές στο γονίδιο C9ORF72 και το υπόλοιπο ποσοστό οφείλεται σε μεταλλαγές σε γονίδια όπως ALS2, SETX, SPG11, VAPB, ANG, OPTN και πιθανόν και σε άλλα.
Κλινικά σημάδια και συμπτώματα
Συσπάσεις, κράμπες και δυσκαμψία μυών
Ατροφία μυών και αδυναμία στα χέρια ή/και στα πόδια
Δυσκολία στην βάδιση
Ατροφία των μυών του προσώπου, της γλώσσας και του αυχένα
Δυσκολία στην ομιλία, στη μάσηση κα στην κατάποση ή ακόμη και στην αναπνοή
Παράλυση
Η νοητική λειτουργία και οι αυτόνομοι μύες όπως οι σφικτήρες και η καρδιά δεν επηρεάζεται.
Ποια είναι η πορεία της νόσου ;
Τα αρχικά συμπτώματα της νόσου είναι μυϊκή αδυναμία και κράμπες και, πιο σπάνια, δυσκολία στην κατάποση και την ομιλία (στην bulbar onset μορφή). Σταδιακά, το άτομο δεν μπορεί να περπατήσει ή να χρησιμοποιήσει τα χέρια του, αδυνατεί να καταπιεί και να μιλήσει. Τέλος χάνεται η λειτουργία των αναπνευστικών μυών και οδηγείται σε αναπνευστική ανεπάρκεια, που είναι και το τελικό στάδιο της νόσου.
Η εξέλιξη της νόσου είναι πιο αργή σε άτομα κάτω των 40 ετών, ελαφρώς παχύσαρκα και με συμπτώματα που περιορίζονται ή ξεκινάνε από τα άκρα. Αντίθετα, άτομα με bulbar-onset και respiratory-onset ALS έχουν κακή πρόγνωση. Η λειτουργία της κύστης και η εντερική κινητικότητα παραμένει άθικτη στην πλειοψηφία των ασθενών μέχρι τα τελικά στάδια της νόσου.
Στο τελευταίο στάδιο της νόσου, η αδυναμία κατάποσης οδηγεί σε εισροφήσεις και αυξάνει τον κίνδυνο πνευμονίας από εισρόφηση. Το άτομο υποσιτίζεται λόγω δυσφαγίας και αδυναμίας των μυών του οισοφάγου. Τέλος, πλήττονται οι αναπνευστικοί μύες και ο ασθενής οδηγείται στον θάνατο. Οι κυριότερες αιτίες θανάτου στα άτομα με ALS είναι η αναπνευστική ανεπάρκεια και η πνευμονία. Τo προσδόκιμο ζωής τυπικά δεν υπερβαίνει τα 5 έτη.
Πώς γίνεται η διάγνωση της ALS ;
Η διάγνωση τίθεται κατά βάση κλινικά, με βάση τα συμπτώματα του ασθενούς. Λαμβάνεται ατομικό και κληρονομικό ιστορικό και γίνεται πλήρης νευρολογική εξέταση προκειμένου η νόσος να διαφοροδιαγνωστεί από άλλες μυϊκές/νευρολογικές παθήσεις. Η διάγνωση επιβεβαιώνεται με ένα ηλεκτρομυογράφημα.
Πώς αντιμετωπίζεται ;
Δεν υπάρχει ακόμα οριστική θεραπεία για την ALS, μόνο επιβράδυνση της πορείας της νόσου, ανακούφιση των συμπτωμάτων και βελτίωση της ποιότητας ζωής των ασθενών. Πιο ειδικά, προσφέρεται φαρμακευτική αγωγή, φυσιοθεραπεία και εργοθεραπεία, διατροφική και αναπνευστική υποστήριξη και βοηθητικές συσκευές για ομιλία και κίνηση.
Η ζωή των ατόμων με ALS είναι ιδιαίτερα δύσκολη και ειδικά στα τελικά στάδια της νόσου, είναι απαραίτητη ή συνεχής φροντίδα και στήριξη από το οικογενειακό περιβάλλον και τους επαγγελματίες υγείας.
zougla.gr




























 Facebook
Facebook Twitter
Twitter Youtube
Youtube RSS Feed
RSS Feed